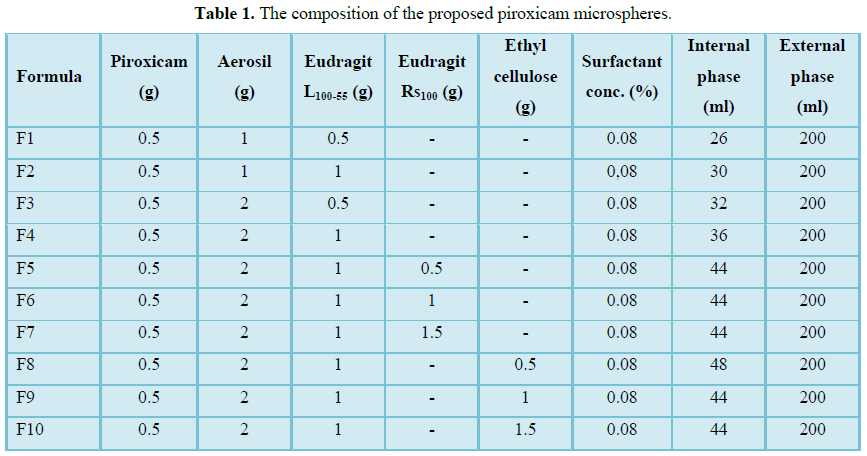
910
Views & Citations10
Likes & Shares
The aim of the present study is to reduce the ulcerogenic effect of
piroxicam by controlling its dissolution rate. Piroxicam is a class II drug
according to BCS, thus dissolution is the rate limiting step for its
bioavailability. To control the bioavailability of piroxicam and reduce its
side effects dissolution rate was attempted to be controlled by preparing
microspheres having a solid dispersion structure. Two different polymers were
used one is solid dispersing polymer to enhance dissolution rate of piroxicam
and the other is a retarding polymer in order to control its release. Depending
on the ratio of the two polymer combinations, drug release can be controlled.
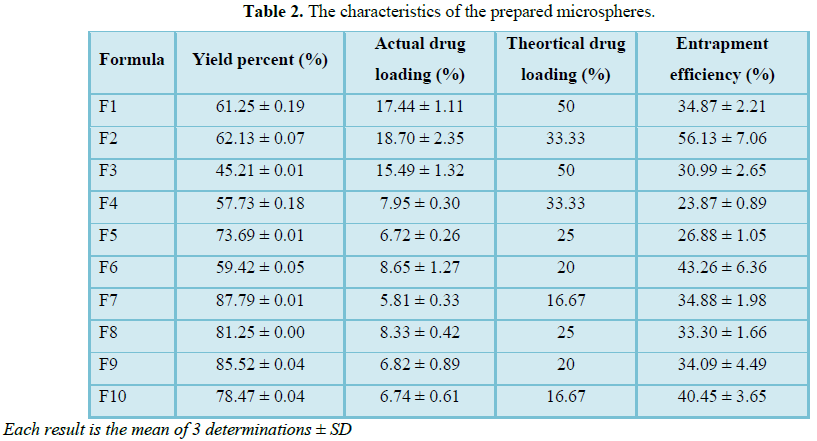
Percentage yield and entrapment efficiency of prepared formulations ranges from
45.21% ± 0.01 to 87.79% ± 0.01 and 23.87% ± 0.89 to 56.13% ± 7.06, respectively
depending on polymer concentration. Characterization of piroxicam and other
formulations using DSC and FTIR analysis reflects possibility of transformation
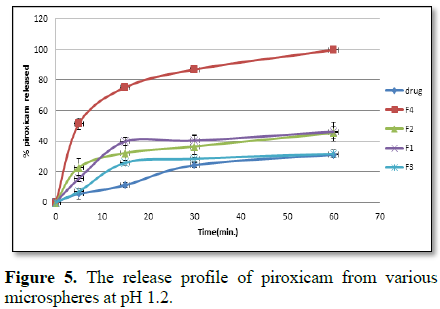
of the drug from crystalline to amorphous state. Release of piroxicam was
faster from microspheres having solid dispersion structure (about 74.98% ± 1.5 after
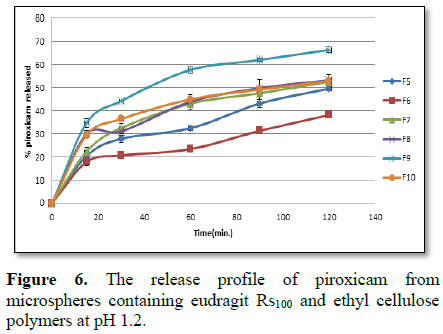
15 min). In order to control the release of the drug, ethyl cellulose as well
as eudragit Rs100 was added. The release pattern of piroxicam from different
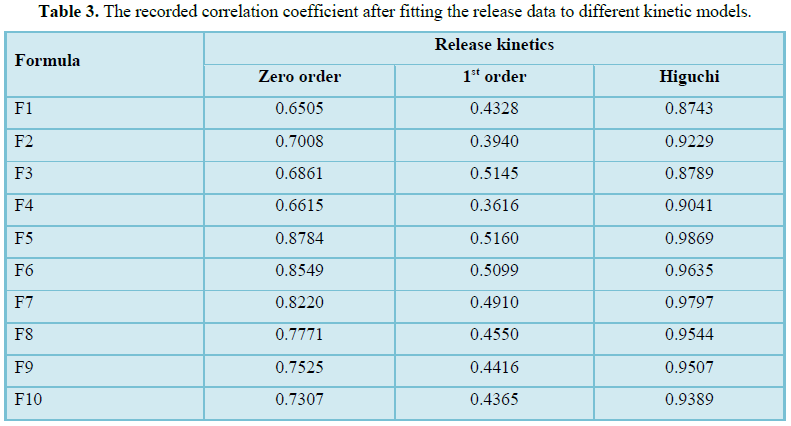
prepared formulations followed Higuchi matrix kinetic model. In vivo
ulcerogenic studies revealed that piroxicam containing eudragit S100 was the
formula of the least ulcer incidence (50%) showing gastric mucosa with (mild
mucosal edema as well as minimal sloughed area and also minimal vascular
congestion) than those showed by other animals.
Keywords: Ulcerogenic,
Piroxicam, Polymers, Microspheres
INTRODUCTION
The permeability besides the solubility behavior of a
drug is a key determinant of its oral bioavailability [1]. Formulation of
poorly soluble compounds for oral delivery now presents one of the interesting
challenges to formulation scientists in the pharmaceutical industry where more
than 40% new chemical entities are practically insoluble [2].
Piroxicam is a member of the oxicam group of
non-steroidal anti-inflammatory drugs (NSAIDs) that is indicated for acute or
long-term use in the relief of signs and symptoms of osteoarthritis and
rheumatoid arthritis and also has gastrotoxic as well as duodenotoxic effects
[3,4].
According to the Biopharmaceutical Drug Classification
System (BCS) piroxicam is a class II drug, characterized by low solubility-high
permeability, where drug dissolution is the rate limiting step in drug
absorption and bioavailability [5].
Solid dispersion systems in which the drug is dispersed
in solid water-soluble matrices either molecularly or as fine particles have
shown promising results in increasing bioavailability of poorly water-soluble
drugs [6,7]. Solid dispersion techniques including dissolution method, fusion
method and fusion-dissolution method were commonly used.
Microencapsulation
for oral use has been employed to sustain the drug release and to reduce or
eliminate gastrointestinal tract irritation. In addition, multiparticulate
delivery systems spread out more uniformly in the gastrointestinal tract. Small
particle size, are widely distributed throughout the gastrointestinal tract
which improves drug absorption and reduces side effects due to localized
build-up of irritating drugs against the gastrointestinal mucosa [10,11].
Emulsion solvent
evaporation method was used for the preparation of microspheres due to its ease
of fabrication without compromising the activity of drug and it requires only
mild conditions such as ambient temperature and constant stirring [12,13].
Piroxicam may cause
serious GI side effects, including ulceration as well as intestine and stomach
perforation, which can also be fatal [14,15]. Piroxicam side effects are
similar to other NSAIDs, its GI damage is the most serious one (GI adverse
effects is 3.7-10 fold) [16].
Attempts to overcome
the undesired effects of piroxicam include modifications in the manner of
administration [17-19]; in pharmaceutical forms [20], in the preparation of
pro-drugs [21]; and in the synthesis of complexes [22].
The aim of the
present study is preparation of piroxicam microspheres with different polymers
to obtain different dissolution patterns and comparing it’s in vivo gastro ulcerogenic activity with
free piroxicam and piroxicam microspheres containing eudragit S100 which was
previously prepared by El-Kayad et al. [23].
MATERIALS AND METHODS
Materials
Piroxicam was
obtained as a gift sample from Medical Union Pharmaceuticals, Ismailia, Egypt.
Ethyl cellulose, Eudragit Rs100 and Eudragit L100-55 were obtained as gift
samples from Sigma for Pharmaceutical Industries, Quesna, Egypt. Aerosil
(ISO-CHEM, China). Ethanol, methanol, dichloromethane, sodium lauryl sulphate
(pharmaceutical grade) were obtained from El Nasr pharmaceutical chemicals
company, Cairo, Egypt. All other chemicals used were of analytical grade.
Equipment
Mechanical paddle
stirrer (Heidolph RZR-2000), U.V. visible spectrophotometer (Shimadzu
UV-visible UV-160 A, Japan), USP II dissolution apparatus (paddle type, Copley
Scientific Dis 6000, Nottingham, UK).
Determination of piroxicam by UV-visible
spectrophotometric method
A stock solution of
piroxicam in methanol (1000 µg/ml) was prepared. The standard stock solution
was further diluted to the required concentration for method development and
validation. Calibration curve was constructed at different pH values (1.2, 6.8
and 7.4) using 0.1 N HCl and phosphate buffer, respectively. Ultraviolet
absorbance of the solutions was determined spectrophotometrically (Thermo,
Evo300pc, USA) at the wavelength of maximum absorbance at 334, 354 and 353 nm
for pH values 1.2, 6.8 and 7.4, respectively [24].
Preparation of piroxicam microspheres
Surface morphology (SEM): The surface
morphology and texture of the prepared microspheres were determined using
scanning electron microscope (SEM). A small amount of each sample was spread on
aluminum stub and coated with gold then placed in SEM chamber using SEM
(JEOL-JSM-5200 LV, Japan). SEM photomicrograph was taken at acceleration
voltage of 25 KV.
Percentage-yield: The prepared microspheres
were collected after drying and weighed [27]. Percentage yield of the
microspheres was calculated as follow:
% yield of prepared microspheres = (actual weight of
the product/total weight of excipients and drug) × 100
Fourier transformed infrared spectroscopy (FT-IR): Interaction between drug and polymers was investigated using IR
spectrophotometer. IR spectroscopy was performed using Fourier- transform
infrared spectrophotometer, (Jasco, Japan). Eudragit L100-55, eudragit Rs100,
ethyl cellulose, aerosil, piroxicam, prepared formulations and physical mixture
between drug and different polymers spectrum were recorded using FTIR spectrophotometer.
Samples were mixed with potassium bromide (spectroscopic grade) and compressed
into disks using hydraulic press before scanning between 4000 and 400 cm-1
at a resolution of 4 cm-1 [28].
Entrapment efficiency: The entrapment efficiency
(%) of the prepared microspheres was evaluated using the method of Gangadhar et
al. [29]. with certain modification. 25 mg of each prepared formula were
crushed into powder and were completely dissolved in 100 ml of phosphate buffer
solution (pH 7.4) using magnetic stirrer. 5 ml of the obtained solution was
filtered using syringe filter (0.45 µm) and the concentration of the drug was
determined spectrophotometrically at 353 nm after appropriate dilution [30,31].
The actual drug loading and encapsulation efficiency (EE %) were calculated
using the following equations:
Encapsulation efficiency (%) = (Actual drug
loading/Theoretical drug loading) × 100
Differential scanning calorimetry (DSC): DSC studies were performed using a DSC Perkin Elmer with thermal
analyzer. A known weight of the test sample was loaded in aluminum pans which
were crimped and mounted on the DSC before heating under nitrogen flow (20
ml/min). Thermal results were recorded while heating from 30 to 400°C at a
heating rate of 10°C/min. An empty aluminum pan was used as a reference. DSC
thermograms of pure substances, their physical mixture and drug loaded
microspheres were recorded.
In vitro drug release study
In vitro drug release from the prepared microspheres was performed at different
pH values (1.2 and 6.8) at 37 ± 0.5°C. The release of piroxicam from
microspheres was determined using type II dissolution apparatus (Copley, NG
42JY, Nottingham, UK). Microspheres equivalent to 20 mg were weighed and added
to 900 ml of dissolution medium with a stirring rate of 100 rpm. For
microspheres having solid dispersion structure release was measured at pH 1.2
for 1 h. The pH of the dissolution medium was kept at 1.2 for 2 h then adjusted
to 6.8 for 4 h to evaluate release of piroxicam from microspheres containing
eudragit Rs100 and ethyl cellulose. Samples (5 ml) were withdrawn from the
dissolution medium at various time intervals and replaced with 5 ml fresh media
to keep sink conditions. The amount of drug released at each time interval was
calculated and the cumulative amount of drug released was calculated as a
function of time to construct the drug release profile.
Release kinetics studies
To determine the possible release mechanism of
different prepared formulations the release data was fitted to different
kinetic models. Thus, the release data was fitted to zero order, first order
and Higuchi kinetic models [32].
In vivo ulcerogenicity
studies
Animals, treatment and collection of tissue samples:
Male Wistar-strain rats weighing (160-180) g were obtained from National
researches center (Cairo, Egypt). In vivo ulcerogenicity studies were conducted
according to the procedure reported by previous study with some modifications
[33].
Animals were maintained at 22 ± 1°C with 12 h
light/dark cycle using galvanized wire cages and allowed rat chow and water ad libitum for 14 days to get adapted to
laboratory conditions. In vivo
experimental protocols were approved by the Animal Care and Use Committee and
were in accordance with all recommendations in the University Guide for the
Care and Use of Experimental Animals.
The animals were divided into four groups each
containing 6 animals (n=6). Animals were fasted 40 h with free access to water
[34]. The first group of animals is the control group, the second group of animals
was treated with free piroxicam (30 mg/kg), the third group of animals was
treated with piroxicam microspheres containing eudragit S100 in the ratio (1:3)
in a dose equivalent to (30 mg/kg) of piroxicam while the fourth group of
animals was treated with piroxicam microspheres containing aerosil and eudragit
L100-55 in the ratio (1:4:2) in a dose equivalent to (30 mg/kg) of
piroxicam.
Piroxicam and prepared formulae were administrated
orally to each corresponding group as 1 ml suspension by oral gavage using an
intubation needle fitted onto a syringe of appropriate size in a dose
equivalent to 30 mg/kg of piroxicam or its equivalent in different formulations
[35,36].
6 h later, each animal was removed from its cage,
anaesthetized with ether and the abdomen was opened. Each stomach was excised,
dissected along the greater curvature and contents were emptied by gently
rinsing with isotonic saline solution [37].
Macroscopic examination of gastric ulcers: After the
animals were sacrificed, each stomach was pinned out on a flat surface with the
mucosal surface uppermost. Then a 10x binocular magnifier was used to examine
and assess presence of hemorrhagic lesions and/or gastric ulcers expressed as
the ulcer incidence.
The number of erosions per stomach was assessed for
severity according to the scoring system described [38]. The grade of lesions
was scored according to the following scale- 0: no pathology; 1: small (1-2 mm
ulcers); 2: medium (3-4 mm ulcers); 4: large (5-6 mm ulcers); 8: ulcers
(greater than 6 mm). The sum of the total ulcer scores in each group of rats
was divided by the number of animals in the group to give the mean ulcer index
for that group.
Histopathological examination of stomach sections: The collected stomachs samples were fixed overnight in 10% w/v buffered
formalin. Each specimen was sectioned, processed overnight and then embedded in
paraffin. The paraffin blocks were sectioned and the slides were stained with a
standard haematoxylin and eosin stain then photographed under 20x magnifications
using a Nikon Eclipse 80i light microscope (Nikon Corporation, Japan) [39].
RESULTS AND DISCUSSION
Surface morphology
The percentage yield of different formulations was
represented in Table 2 ranging from 45.21 ± 0.01 to 87.79 ± 0.01. The
percentage yield of microspheres having solid dispersion structure is less than
those containing controlling release polymers. Formula F3 and formula F4 having
high percentage of aerosil has been found to have the least yield. This can be
attributed to that aerosil have high porosity and specific surface area to act
as dispersing agent may cause loss of the drug. By increasing polymer amount,
percentage yield of the obtained formulations is increased. This was approved
by work of other researchers who study the effect of the polymer concentration
on the percentage yield of the resulting microspheres [40].
Entrapment efficiency varies according to polymer
type, drug to polymer ratio and aerosil percentage as shown in Table 2.
Effect of aerosil on entrapment efficiency is due to the fact that aerosil
particles have high porosity and large specific surface area leading to drug
loss during evaporation of organic solvent within the preparation process. Thus
increasing amount of aerosil decreases entrapment efficiency as shown for
formula F2 which has the highest entrapment efficiency (about 56%) having the
least amount of aerosil and the highest amount of eudragit L100-55. Increasing
polymer percentage increases the entrapment efficiency due to better coating of
drug resulting from precipitation of polymer on the surface of the dispersed
phase which leads to preventing of drug diffusion across the phase boundary
[41]. Similar results were obtained by Mehta et al. [42] and Sharma et al. [43].
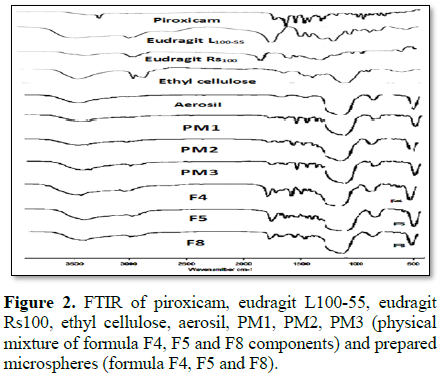
Fourier transformed infrared spectroscopy (FTIR)
Accordingly, the results ruled out the possibility
of disappearance of intramolecular hydrogen bonding as 1632 cm-1
stretching peak which is involved in the formation of this intramolecular
hydrogen bond shifted to higher value 1642 cm-1. For IR spectrum of
microspheres containing eudragit Rs100 presence of 1527 cm-1, 1601
cm-1, 1329 cm-1 peak may be due to intermolecular
interaction between drug and polymer. The same results were obtained by other
investigators studying interaction between piroxicam and eudragit polymers
[47]. Physical mixture spectrum indicates only the summation of different
components of microspheres.
Differential scanning calorimetry (DSC)
According to biopharmaceutical classification system
piroxicam is a class II drug having low solubility and high permeability so
drug release is a crucial and a limiting step for oral drug bioavailability
particularly for drugs with low gastrointestinal solubility and high
permeability. By improving the drug release profile of these drugs it is
possible to enhance their bioavailability and reduce side effects [52].
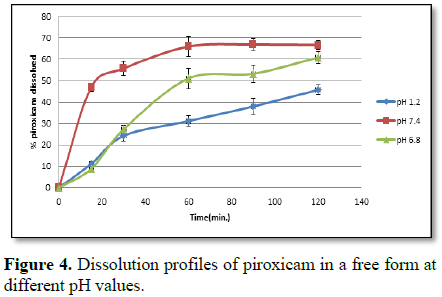
As piroxicam has both acidic and basic groups, its
solubility is pH dependent so the difference in the degree of the dissolution
of the drug is dependent on the ionization of the drug at different pH values
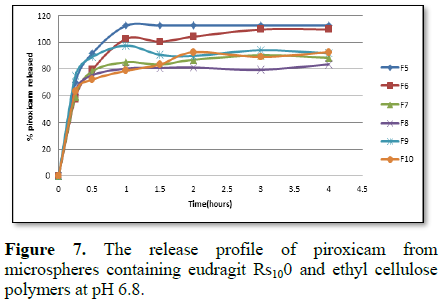
[53]. Release of the drug at different pH values is illustrated in Figure 4.
In vivo ulcerogenicity
studies
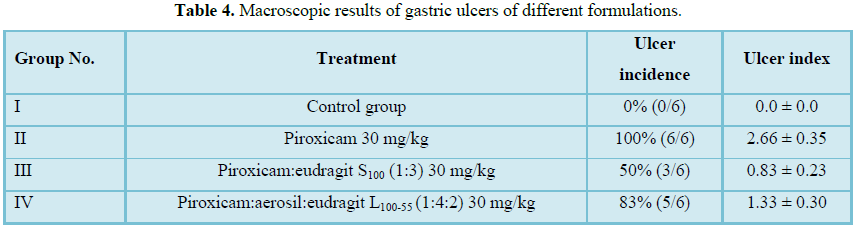
Macroscopic analysis: Experimental design, animal groups as well as ulcer incidence and ulcer index of different formulations are illustrated in Table 4. Figure 8 shows macroscopic observations of stomach mucosa of the animals of different groups which differ according to presence or absence of hemorrhagic lesions.
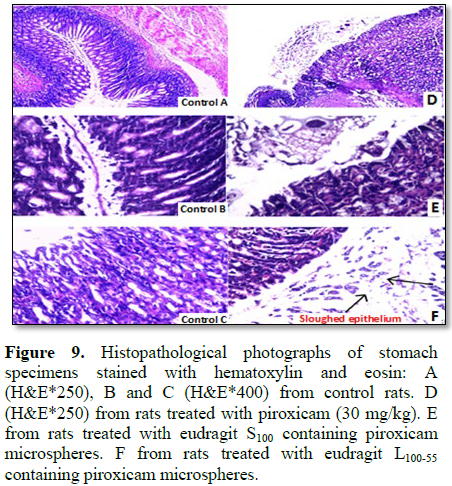
Histopathological analysis: The results of the
histopathological analysis of different stomach specimens of the animals of
different groups after investigation under microscope are illustrated in Figure
9.
Piroxicam treated rat (group II) represented by Figure
9D show stomach mucosa of gastro-esophageal junction of the treated group
with piroxicam (30 mg/kg) after 6 h having focal superficial degeneration,
congestion and sloughing of gastric mucosa with wide inflammatory cellular
infiltration and dense mononuclear cell infiltration, respectively.
Figure 9E (group III) show stomach mucosa of animals
treated with microspheres containing piroxicam and eudragit S100
(equivalent to 30 mg piroxicam/kg) after 6 h. It shows gastric mucosa having
(mild mucosal edema with minimal sloughed area and vascular congestion).
Figure 9F (group IV) show stomach mucosa of animals
treated with microspheres containing piroxicam and eudragit L100-55
(equivalent to 30 mg piroxicam/kg) after 6 h. It shows superficial diffuse,
sloughing of the covering epithelium, severe congestion and inflammatory
cellular infiltration of (group IV). So according to the obtained results it
was found that group III of microspheres containing eudragit S100
and piroxicam was the group of decreased in vivo gastric ulcerogenic activity compared
to other groups.
CONCLUSION
1. Leuner
C, Dressman J (2000) Improving drug solubility for oral delivery using solid
dispersions. Eur J Pharm Biopharm 50: 47-60.
2. Patel
RC, Keraliya RA, Patel MM, Patel NM (2010) Formulation of furosemide solid
dispersion with micro crystalline cellulose for achieving rapid dissolution. J
Adv Pharm Tech Res 1: 180-189.
3. (2002)
Physicians’ Desk Reference. 56th Edn. Medical Economics Company,
Inc.: Montvale N.J., p: 2685.
4. Brune
K, Dietzel K, Nurnberg B, Schneider HT (1987) Recent insight into the mechanism
of gastrointestinal tract ulceration. Eur J Rheumatol Inflamm 9: 8-14.
5. Amidon
GL, Lennernäs H, Shah VP, Crison JR (1995) A theoretical basis for a
biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res 12: 413-420.
6. Chiou
WL, Riegelman S (1971) Pharmaceutical applications of solid dispersion systems.
J Pharm Sci 60: 1281-1302.
7. Ford
JL (1986) The current status of solid dispersions. Pharm Acta Helv 61: 69-88.
8. Bansole
SS, Banerjee SK, Gaikward DD, Jadhav SL, Thorat RM (2010) Microencapsulation: A
review. Int J Pharm Sci Rev Res 1: 38-43.
9. Obeidat
WM (2009) Recent patients review in microencapsulation of pharmaceuticals using
emulsion solvent removal methods. Recent Patent Drug Deliv Formul 3: 178-192.
10. Sam
TM, Gayathri SD, Prasanth VV, Vinod B (2008) NSAIDs as microspheres. Internet J
Pharmacol 6.
11. Li
SP, Kowalski CR, Feld KM, Grim WM (1988) Recent advances in microencapsulation
technology and equipment. Drug Dev Ind Pharm 14: 353-376.
12. Basu
SK, Adhiyaman R (2008) Preparation and characterization of nitrendipine loaded
eudragit RL 100 microspheres prepared by an emulsion-solvent evaporation
method. Trop J Pharm Res 7: 1033-1041.
13. Lakshmana
PS, Shirwaikar AA, Shirwaikar A, Kumar A (2009) Formulation and evaluation of
sustained release microspheres of rosin containing aceclofenac. Ars Pharm 50:
51-62.
14. Piroxicam official FDA information (2010) Side
effects and uses. Available at: http://www.drugs.com/pro/piroxicam.html
15. Giercksky
KE (1986) Piroxicam and gastrointestinal bleeding. Am J Med 81: 2-5.
16. Rodr´ıguez
LA, Hern´andez-D´ıaz S (2001) Relative risk of upper gastrointestinal
complications among users of acetaminophen and non-steroidal anti-inflammatory
drugs. Epidemiology 12: 570-576.
17. Ligumsky
M, Sestieri M, Karmeli F, Zimmerman J, Okon E, et al. (1990) Rectal
administration of non-steroidal anti-inflammatory drugs. Gastroenterology 98:
1245-1249.
18. Benko
S, Ezal G, Nagy E, Klebovich O (1992) Comparative bioavailability of two rectal
preparations of piroxicam in man. Eur J Clin Pharmacol 43: 315-317.
19. Mueller
BA, Rex DK, Figueroa N, Greene P, Brater DC (1992) A pharmacokinetic and
endoscopic comparison of an oral and an experimental buccal piroxicam
formulation. Pharmacotherapy 12: 93-97.
20. Aabaken
L, Olaussen B, Mowinckel P, Osnes M (1992) Gastroduodenal lesions associated
with two piroxicam formulations. An endoscopic comparison. Scand J
Gastroenterol 27: 1049-1054.
21. Esteve
J, Farre AJ, Roser R (1986) Pharmacological profile of droxicam. Gen Pharmacol
8: 423-429.
22. Cadel
S, Bongrani S (1990) Beta-cyclodextrin complexation improves absorption and
gastric tolerability. Acta Physiol Hung 75: 45-46.
23. Zein
EE, Donia AA, El-Kayad S (2016) Microencapsulation of piroxicam using pH
sensitive polymers. Eur J Pharm Med Res 3: 74-80.
24. Karatas
A, Yüksel N, Baykara T (2005) Improved solubility and dissolution rate of
piroxicam using gelucire 44/14 and labrasol. Farmaco 60: 777-782.
25. Mao
S, Shi Y, Li L, Xu J, Schaper A, et al. (2008) Effect of process and
formulation parameters on characteristics and internal morphology of poly (D,
L-lactide-co-glycolide) microspheres formed by the solvent evaporation method.
Eur J Pharm Biopharm 68: 214-223.
26. Cui
F, Yang M, Jiang Y, Cun D, Lin W, et al. (2003) Design of sustained-release
nitrendipine microspheres having solid dispersion structure by quasi-emulsion
solvent diffusion method. J Control Release 91: 375-384.
27. Chandiran
IS, Sivakumar T, Kumar BP (2010) Preparation and evaluation of aceclofenac
loaded biodegradable microspheres. Int J Pharm Biomed Res 1: 19-23.
28. Essa
EA, Elkotb FE, Zin Eldin EE, El-Maghraby GM (2015) Development and evaluation
of glibenclamide floating tablet with optimum release. J Drug Deliv Sci Technol
27: 28-36.
29. Gangadhar
CB, Sunder SR, Vimal KV, Raju SM, Kiran SM (2010) Formulation and evaluation of
indomethacin microspheres using natural and synthetic polymers as controlled
release dosage forms. Int J Drug Dis 2: 8-16.
30. Abdallah
MH, Sammour OA, El-Ghamry HA, El-Nahas HM, Barakat W (2012) Development and
characterization of controlled release ketoprofen microspheres. J Appl Pharm
Sci 2: 60-67.
31. Yerriswamy
B, Reddy CLN, Prasad CV, Subha MCS, Rao KC (2010) Controlled release studies of
5-fluorouracil through poly (vinyl caprolactum-co-vinyl acetate) microspheres.
Asian J Pharm 4: 200-204.
32. Costa
P, Lobo JMS (2000) Modeling and comparison of dissolution profiles. Eur J Pharm
Sci 13: 123-133.
33. Alsarra
I, Ahmed M, Alanazi F, El-Tahir K, Alsheikh A (2010) Influence of cyclodextrin
complexation with NSAIDs on NSAID/cold stress-induced gastric ulceration in
rats. Int J Med Sci 7: 232-239.
34. El-Shitany
N (2006) Mechanism of omeprazole induced gastric protection against ethanol
induced gastric injury in rats: Role of mucosal nitric oxide and apoptotic cell
death. Proceeding of 1st International Egyptian-Jordanian Conference
on Biotechnology and Sustainable Development: Current Status & Future
Scenarios. Med Pharm 2: 183-193.
35. Bhargava
K, Gupta M, Tangri K (1973) Mechanism of ulcerogenic activity of indomethacin
and oxyphenbutazone. Eur J Pharmacol 22: 191-195.
36. Brzozowski
T, Konturek P, Konturek S (2001) Classic NSAID and selective cyclooxygenase
(COX)-1 and COX-2 inhibitors in healing of chronic gastric ulcers. Microsci Res
Technol 1: 343-353.
37. Chandranath
S, Bastaki S, Singh J (2002) A comparative study on the activity of
lansoprazole, omeprazole and PD-136450 on acidified ethanol- and
indomethacin-induced gastric lesions in the rat. Clin Exp Pharmacol. Physiol
29: 173-180.
38. Bandyopadhyay
D, Ghosh G, Bandyopadhyay A, Reiter RJ (2004) Melatonin protects against
piroxicam-induced gastric ulceration. J Pineal Res 36: 195-203.
39. Haggag
YA, El-Gizawy SA, Zein El-din EE, El-Shitany NA, Osman MA (2016) Sulindac solid
dispersions: Development, characterization and in vivo evaluation of ulcerogenic activity in rats. J Appl Pharm
Sci 6: 22-31.
40. Najmuddin
M, Shelar S, Ali A, Patel V, Khan T (2010) Formulation and in vitro evaluation
of floating microspheres of ketoprofen prepared by emulsion solvent diffusion
method. Int J Appl Pharm 2: 13-17.
41. Rafati
H, Coombes AGA, Adler J, Holland J, Davis SS (1997) Protein-loaded PLGA
microparticles for oral administration: Formulation, structural and release
characteristics. J Control Release 43: 89-102.
42. Mehta
RC, Thanoo BC, De Luca PP (1996) Peptide containing microspheres from low
molecular weight and hydrophilic poly(D, L-lactideco-glycolide). J Control
Release 41: 249-257.
43. Sharma
M, Kohli S, Dinda A (2015) In vitro
and in vivo evaluation of repaglinide
loaded floating microspheres prepared from different viscosity grades of HPMC
polymer. Saudi Pharm J 23: 675-682.
44. Fernandez
M, Rodriguez IC, Margarit MV, Cerezo A (1992) Characterization of solid
dispersions of piroxicam/polyethylene glycol 4000. Int J Pharm 84: 197-202.
45. Mihalic
M, Hofman H, Kuftinec J, Krile B, Caplar V (1986) Piroxicam. Anal Profiles Drug
Subs 15: 509-531.
46. Hao
S, Wang B, Wang Y, Zhu L, Wang B (2013) Preparation of eudragit L 100-55
enteric nanoparticles by a novel emulsion diffusion method. Colloids Surf B
Biointerfaces 108: 127-133.
47. Lin
SY, Lee CJ, Lin YY (1995) Drug-polymer interaction affecting the mechanical
properties, adhesion strength and release kinetics of piroxicam-loaded Eudragit
E films plasticized with different plasticizers. J Control Release 33: 375-381.
48. Kogermann
K, Aaltonen J, Strachan CJ, Pöllänen K, Veski P, et al (2007) Qualitative in
situ analysis of multiple solid-state forms using spectroscopy and partial
least squares discriminant modeling. J Pharm Sci 96: 1802-1820.
49. Vrecer
F, Vrbinc M, Meden A (2003) Characterization of piroxicam crystal
modifications. Int J Pharm 256: 3-15.
50. Tantishaiyakul
V, Kaewnopparat N, Ingkatawornwong S (1999) Properties of solid dispersions of
piroxicam in polyvinylpyrrolidone. Int J Pharm 181: 143-151.
51. Bettinetti
G, Mura P (1994) Dissolution properties of naproxen in combinations with
polyvinyl pyrrolidone. Drug Dev Ind Pharm 20: 1353-1366.
52. Amidon
GL, Lennerna¨s H, Shah VP, Crison JR (1995) A theoretical basis for a biopharmaceutic
drug classification: The correlation of in
vitro drug product dissolution and in
vivo bioavailability. Pharm Res 12: 413-420.
53. Shohin
IE, Kulinich JI, Ramenskaya GV, Abrahamsson B, Kopp S, et al. (2013) Biowaiver
monographs for immediate release solid oral dosage forms: Piroxicam. J Pharm
Sci 1-11.
54. Liu
C, Goud K, Desai H, Tang X, Chen X (2006) Drug release kinetics of spray-dried
chitosan microspheres. J. Drying Technol 24: 769-776.
55.
Babay D, Hoffman A, Benita S (1988)
Design and release kinetic pattern evaluation of indomethacin microspheres
intended for oral administration. J Biomater 9: 482-488.
-
 Table 1
Table 1 -
Table 2
-
Table 3
-
Table 4